Введение
В настоящее время известно несколько десятков заболеваний, связанных с дефектами биогенеза лизосом или их ферментов. Наследственный дефект деградации цепей гликозаминогликанов (ГАГ) (кислых мукополисахаридов) лежит в основе заболеваний, объединенных общим названием мукополисахаридозы (МПС).
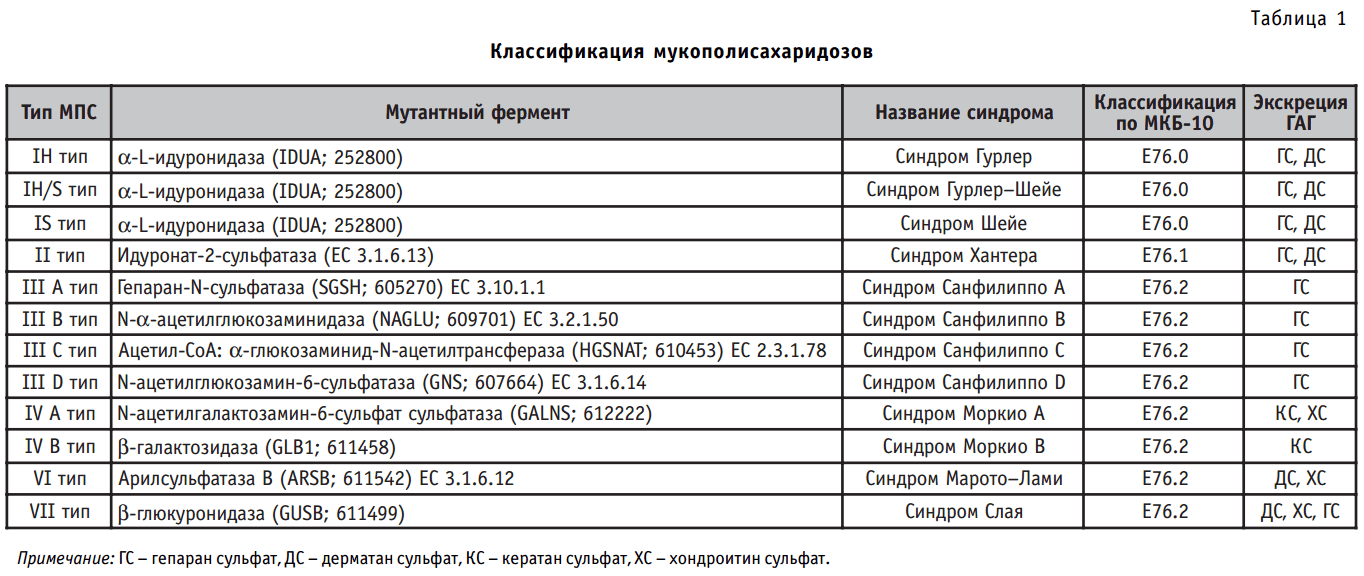
Мукополисахаридозы — группа наследственных болезней обмена веществ, обусловленных дефектом лизосомных гидролаз, участвующих в расщеплении гликозаминогликанов. Нарушение функции фермента вызывает накопление гликозаминогликанов в органах и тканях больного и формирует проявление характерных для мукополисахаридоза фенотипических признаков. Среди мукополисахаридозов выделяют ряд типов, каждый из которых обусловлен дефицитом специфического фермента, участвующего в последовательном расщеплении определенного гликозаминогликана (табл. 1).
Таблица 1. Классификация мукополисахаридозов.
{kind=link}
Мукополисахаридоз типа VI (синдром Марото–Лами, мукополисахаридоз VI, OMIM № 253200) — наследственное заболевание, связанное с дефектом N-ацетилгалактозаминсульфатазы (арилсульфатазы B), одного из пяти ферментов гидролиза важного компонента соединительной ткани дерматан сульфата (ДС). Впервые мукополисахаридоз VI описан в 1960 г. французскими врачами Марото (Р. Maroteaux) и Лами (М. Е. J. Lamy). Частота встречаемости этого заболевания варьируется по данным разных авторов от 1:667 000 до 1:238 000 и составляет в странах Европы 1:434 000. Синдром наследуется по аутосомно-рецессивному типу. Родители являются гетерозиготными носителями патологического гена. Риск повторного рождения больного ребенка в семье составляет 25%.
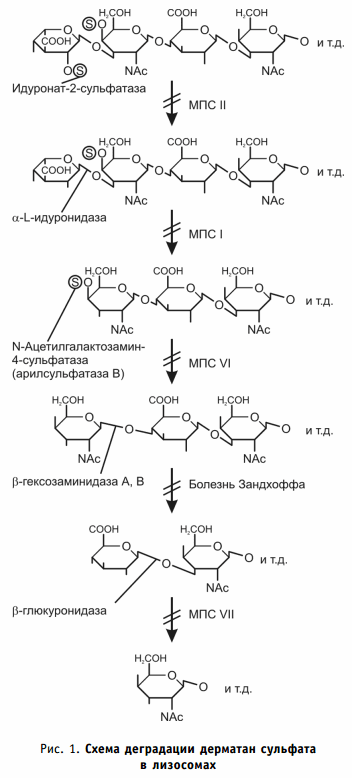
Арилсульфатаза B (ARSB; EC 3.1.6.12) осуществляет гидролиз сульфатной группы N-ацетилгалактозамин-4-сульфата в молекуле дерматан сульфата и сульфатной группы в хондроитин-4-сульфате (рис. 1).
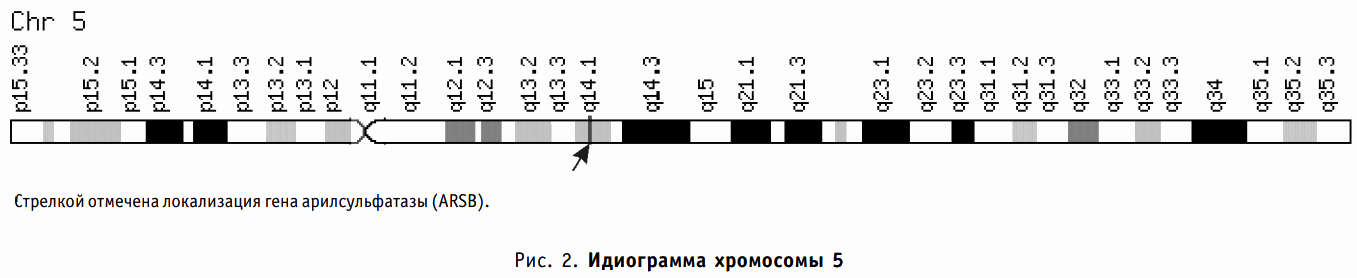
Ген арилсульфатазы B (ARSB) локализуется на хромосоме 5 человека (рис. 2) и включает 7 интронов и 8 экзонов. К настоящему моменту зарегистрировано более 100 мутаций ARSB гена.
Рис. 2. Идиограмма хромосомы 5.
{kind=link}
Мутации гена приводят к накоплению негидролизованного дерматан сульфата внутри лизосом клеток. Дерматан сульфат соединительной ткани является эндотоксином и стимулирует реакцию воспаления, приводит к апоптозу клеток. Поэтому одним из ведущих проявлений мукополисахаридоза является системное поражение всех соединительнотканных образований организма: костей скелета, сухожилий, кровеносных сосудов и клапанов сердца, клеток кожи, воздухоносных путей.
Клинический фенотип мукополисахаридоза VI гетерогенен. Манифестация заболевания отмечается как на первом году жизни с быстрым прогрессированием клинических симптомов, так и во втором десятилетии с относительно легким течением заболевания. Интеллектуальное развитие обычно не страдает. Клиническими симптомами являются постепенное формирование грубых черт лица и скелетных деформаций (множественный дизостоз). Дети с мукополисахаридозом VI в первые годы жизни часто выглядят более крупными, чем их сверстники, однако постепенно становится очевидной задержка роста с остановкой к 6–8 годам, рост больных варьируется в пределах 110–140 см. В этом же возрасте больные обычно начинают страдать частыми инфекциями верхних и нижних дыхательных путей, отитами. Постепенно может развиться шумное дыхание и хронический ринит. Для данного заболевания характерны плотная кожа и умеренно выраженный гирсутизм, густые и жесткие волосы. Нередко выявляются грыжи (пупочная, паховая), гепатоспленомегалия. Отмечаются сгибательные контрактуры суставов верхних конечностей (больные не могут поднять руки вверх, значительно ограничено разгибание в локтевых суставах). С возрастом появляются контрактуры в суставах нижних конечностей. Мукополисахаридоз VI по характеру костных изменений относится к заболеваниям с поражением эпифизов, с чем и связаны трудности дифференциальной диагностики с другими эпифизарными дисплазиями. Рентгенологически наиболее характерны изменения таза, тазобедренных суставов и кистей. Вертлужные впадины широкие, уплощенные, деформированные, крылья подвздошных костей изогнуты, нависают над головками бедренных костей. Наблюдается эпифизарная дисплазия в проксимальной части бедренных костей. Головки гипоплазированы, уплощены, истончены шейки бедренных костей. Возможен асептический некроз головки бедренной кости. В трубчатых костях — нарушение энхондрального и перихондрального окостенения.
В кисти запаздывает появление ядер окостенения костей запястья, пястные кости короткие, широкие, проксимальные их отделы сужены, фаланги пальцев широкие, дистальные фаланги гипопластичны.
При радиологическом исследовании черепа отмечаются деформации, уплотнение костей свода и основания. Черепные швы растянуты, края их истончены. Турецкое седло уплощено, вход в него расширен. Характерны гипоплазия или отсутствие зуба второго шейного позвонка (CII), признаки нестабильности атлантозатылочного сустава, платибазия, компрессия спинного мозга. При исследовании позвоночника выявляются деформации тел позвонков (кубовидная, клиновидная, языкообразная). Возникновение костно-суставных деформаций приводит к развитию чувства парестезии, а впоследствии к боли в суставах, что требует ежедневного приема анальгезирующих лекарственных препаратов. Дети быстро устают в покое, и при физической нагрузке походка постепенно ухудшается, многие пациенты перестают ходить.
Характерным симптомом для мукополисахаридоза VI является прогрессирующее помутнение роговицы, что обусловлено накоплением в ее ткани гликозаминогликанов. Патология органа зрения также включает в себя снижение остроты зрения, гиперметропию и сложный астигматизм различной степени тяжести.
Патогномоничными симптомами заболевания являются комбинированные пороки сердца, среди которых чаще встречается стеноз, недостаточность и регургитация аортального и митрального клапанов, что также связано с отложением гликозаминогликанов в клапанах сердца.
Постепенно формируется клинический фенотип:
- низкий рост,
- короткая шея,
- макроцефалия,
- лицевые дисморфии (увеличение лобных бугров, густые брови, короткий нос с открытыми вперед ноздрями, большой открытый рот, толстые губы),
- макроглоссия,
- гиперплазия десен,
- аномалии числа зубов,
- шумное дыхание,
- диспропорциональное телосложение (укороченное туловище, выступающий живот, выраженный лордоз поясничного отдела, бочкообразная или колоколообразная грудная клетка, вынужденное согнутое положение тела),
- тугоподвижность мелких и крупных суставов,
- «когтистая» кисть,
- сгибательные контрактуры тазобедренных, коленных, локтевых суставов.
Продолжительность жизни при тяжелом течении заболевания не превышает 20 лет, при относительно легком течении заболевания пациенты с мукополисахаридозом VI могут доживать до 40-50 лет.
Диагностика мукополисахаридоза VI основывается на анализе совокупности клинических проявлений, инструментальных и лабораторных данных. Лабораторная диагностика включает проведение биохимических исследований проб крови и мочи по стандартной селективной скринирующей программе диагностики наследственных нарушений обмена. Суммарная экскреция гликозаминогликанов с мочой во всех случаях существенно повышена. Точная идентификация типов мукополисахаридоза возможна путем исследования активности лизосомных ферментов, участвующих в обмене гликозаминогликанов, в лейкоцитах крови, культуре фибробластов кожи, биоптатов печени. При мукополисахаридозе VI значительно снижена активность фермента арилсульфатаза В вплоть до нулевых значений.
Дифференциальную диагностику проводят с другими лизосомными болезнями накопления, характеризующимися сходством фенотипа:
- мукополисахаридозами I типа, II типа, VII типа,
- множественной сульфатазной недостаточностью,
- муколипидозами II и III типа,
- сиалидозом,
- маннозидозом,
- фукозидозом,
- Gm1-ганглиозидозом.
Проведение ДНК-анализа в качестве диагностической процедуры затруднено в связи с преобладанием уникальных мутаций в гене ARSB фермента арилсульфатазы В. Однако исследование молекулярных дефектов, приводящих к развитию заболевания мукополисахаридоза VI, способствует пониманию корреляции «генотип-фенотип».
Лечение больных мукополисахаридозами является симптоматическим и заключается в назначении терапии, способствующей стабилизации патологического процесса в опорно-двигательном аппарате, сердечно-сосудистой системе, центральной нервной системе, паренхиматозных органах, органах зрения и слуха.
Ферментозаместительная терапия мукополисахаридоза VI с применением рекомбинантного фермента арилсульфатазы В человека (rhASB) направлена на улучшение катаболизма гликозаминогликанов, предупреждение накопления их в организме.
Заключение
Мукополисахаридоз VI типа имеет характерные фенотипические проявления, выраженную вариабельность возраста манифестации, а также темпов прогрессирования поражения тканей и органов. Наиболее значимыми клиническими симптомами заболевания являются специфические лицевые дисморфии, патология костно-суставного аппарата, сочетанное поражение митрального и аортального клапанов сердца, помутнение роговицы, снижение остроты зрения. С точки зрения эффективности медико-генетической помощи пациентам с мукополисахаридозом VI особую значимость имеет ранняя постановка диагноза. В этой связи важными научно-практическими задачами исследования мукополисахаридоза VI являются:
- анализ ранних симптомов, которыми манифестирует данный метаболический дефект;
- поиск эффективных фенотипических маркеров;
- изучение клинического полиморфизма.
Кульпанович А. И., Наумчик И. В.
Республиканский научно-практический центр «Мать и дитя».
Журнал «Медицинская панорама» № 9, октябрь 2009.